
UNDERSTANDING TRIGGERS – PREVENTION AND TREATMENT OF SICKLE CELL PAIN CRISIS
SOTA OMOIGUI MD
Board certified Anesthesia and Pain Specialist, L.A. Pain Clinic, Hawthorne, California
Author:
The Biochemical Origin of Pain
Sota Omoigui’s Anesthesia Drugs Handbook
Sota Omoigui’s Pain Drugs Handbook
Website: Medicinehouse.com
Email: [email protected]
PREAMBLE
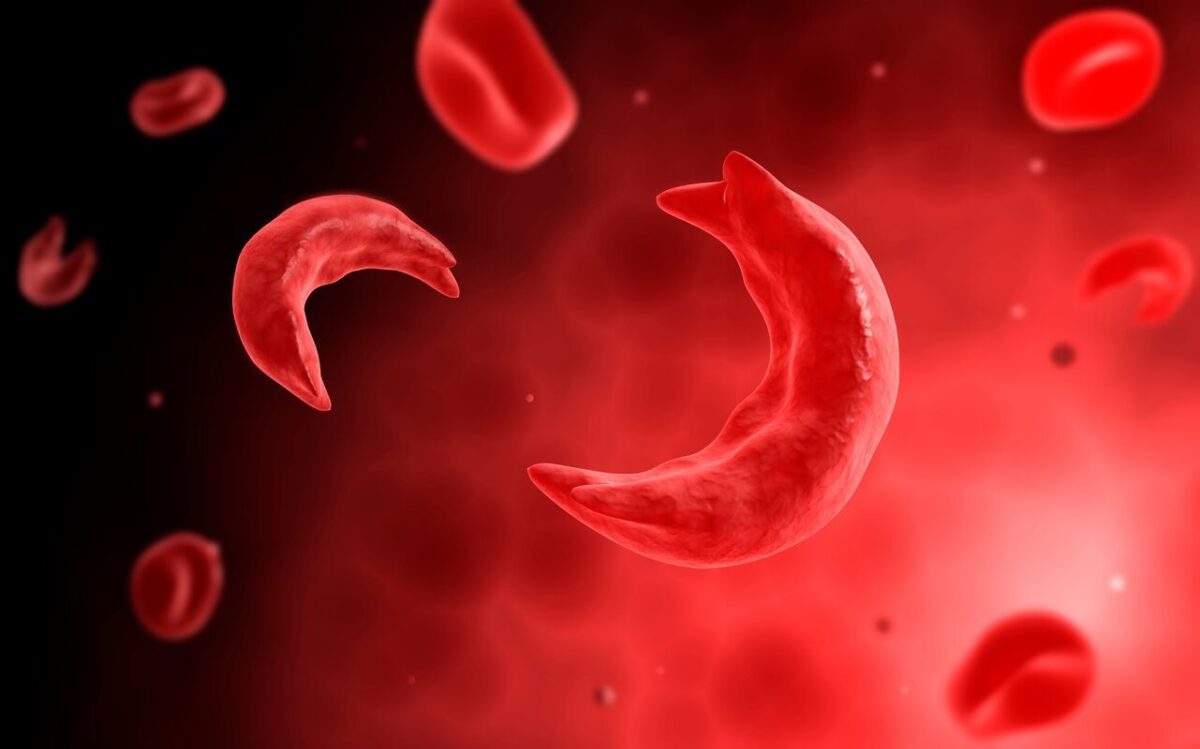
The fundamental defect in sickle cell anemia is the sickling and rigidity of the red blood cells in a low oxygen environment during their passage through the blood capillaries. Science has understood this pathology for the last 50 years, but yet has failed the simplest solution which is to provide Oxygen externally by using an oxygen cylinder or concentrator or at the cellular level by use of the latest medications such as Oxbryta.
It is my goal to bring this knowledge to all those who suffer from and care for patients with sickle cell anemia.
The ultimate vindication of this solution that I have advocated for the last few years is the new medication Oxbryta that is nothing short of a breakthrough treatment and addresses this fundamental defect in sickle cell anemia. With Oxbryta, oxygen is now provided at the cellular level, during the red cell passage through the capillaries, and prevents the sickling and rigidity as well as the hemolysis and anemia.
SICKLE CELL CRISIS AND PAIN TRIGGERS
Pain crisis are predictable and can be avoided as long as you know the triggers. Triggers that contribute to pain crisis include low oxygen saturation, daytime exertion, waking up earlier with a shortened duration of sleep, stress, fatigue, exercise, exposure to cold, ingestion of alcohol, airline travel, altitude that exceeds 2,000 feet, infection, malaria, or pregnancy.
In patients with sickle cell disease (SCD), pain crisis tends to occur most often at night, due to the relative lack of oxygen as a result of varying degrees of sleep apnea, or due to a trigger the patient may have been exposed to during the day. During sleep, minimum oxygen saturation is significantly and may drop from 93.3% +/- 0.4% during wakefulness to 86.5% +/- 0.9%.
This nocturnal lack of oxygen is a prelude to vaso-occlusive crisis. Low nocturnal oxygen saturation is highly significantly associated with a higher rate of painful crisis.”
Other triggers of crises that involve varying degrees of tissue hypoxia may include exercise, fatigue, infection, and exposure to cold. Exposure to cold results in vasoconstriction and delayed transit time, which can trigger a crisis.
Subsequent to commercial airline flights, patients with SCD are known to experience complications such as bone pain, splenic infarction,30,31 osteonecrosis (avascular necrosis) of the hip, and, in some cases, prolonged crisis resulting in death (anecdotal report). These complications have been linked to prolonged decrease in oxygen saturation at high altitudes, with oxygen saturations measured as low as 77%
Preventing a Sickle Cell Crises
Voxelotor (Oxbryta)
This medication increases the affinity of hemoglobin for oxygen, resulting in a decreased concentration of deoxygenated sickle hemoglobin, thereby reducing the amount of red blood cell destruction, and increasing hemoglobin levels.38,39 Hemoglobin response may occur as early as a few days and in some cases return to near-normal levels.
Clinical trial findings40,41 showed that Oxbryta raised hemoglobin levels in 51% of the 90 patients treated with Oxbryta at a high dose of 1500 mg daily, compared with 6.5% of those on placebo. Hemoglobin levels rose over the 24 weeks to a mean of 9.8 g/dL in the patients given Oxbryta at that high dose, and to 8.9 g/dl in 92 patients treated with a lower 900 mg daily study dose. In some patients, hemoglobin may rise as high as 12 g/dl at the high dose.
At high dose, Oxbryta’s use also lowered levels of two established biomarkers of hemolysis: reticulocytes (by 19.9%) and bilirubin (by 29.1%). Oxbryta is administered orally, once daily for patients 12 years and older.
Oxbryta is an effective replacement for blood transfusion to treat anemia in a non-emergent situation as it can raise hemoglobin levels within just a few days.
High dosing of Oxbryta may increase hemoglobin up to 12 g/dl and may be associated with more frequent pain crisis. The dose should be maintained or titrated down to 1500 mg two or three times a week to maintain an optimal hemoglobin concentration of 10 g/dl.
Oxygen
During sleep, decrease in oxygen levels will result in a pain crisis.. Oxygen therapy is a simple yet cost effective way to prevent VOC and its attendant morbidity and mortality.
Prior to sleep or at bedtime, where the oxygen saturation is at or below 90%, and especially in the presence of VOC, triggers such as daytime exertion, shortened duration of sleep, stress, fatigue, exercise, exposure to cold, ingestion of alcohol, airline travel, altitude that exceeds 2,000 feet, infection, and malaria, oxygen should be administered by nasal canula at a rate of 1.5 to 2 liters/minute.
This should be delivered by an oxygen cylinder or preferably by a home or portable oxygen concentrator, to maintain an oxygen saturation of > 95%. This can result in a decrease by 85% to 90% in the frequency of nocturnal vaso-occlusive crises.
Upon onset of a crisis, oxygen administration within the first few minutes may reverse the sickling and terminate the crisis. Subsequently, oxygen is no longer preventive but should be part of an abortive protocol.
Oxygen may be obtained from oxygen cylinders or, more conveniently, and with less maintenance, from portable oxygen concentrators such as the SeQual Eclipse, Inogen, Respironics, AirSep, DeVilbiss, and others, which extract oxygen from ambient air.
Oxygen should be provided in continuous flow from a tank or concentrator. Pulse dose oxygen delivery triggered by inspiratory effort does not provide adequate oxygen delivery and should not be used.
As a matter of public policy, commercial airlines should be mandated to provide medical oxygen to passengers who require it. Continuous flow oxygen could be provided from a piped oxygen supply, an FAA-approved oxygen tank, or an FAA certified concentrator such as the SeQual Eclipse.
See also, Dr. Omoigui’s prior paper on oxygen guidelines in sickle cell crisis.
Hydroxyurea
Hydroxyurea increases production of hemoglobin F, thereby reducing the incidence of sickling. Hydroxyurea has been shown to decrease the rate of painful crises in some patients, related to the size of the HbF treatment response.42
Recommended Dosing: 15 mg/kg/day to start and increase as needed by 5 mg/kg/day every 12 weeks, if blood counts are within acceptable range; Max dose 35 mg/kg/day.
Adakveo
Crizanlizuman-tmca (Adakveo)is a monoclonal antibody targeted against the P-selectin glycoprotein that is expressed on activated endothelial cells and platelets. It can be considered for use as part of a comprehensive management of a pain crisis.43
Blood Transfusion
Due to the risks of alloimmunization and hemolytic transfusion reaction, the routine use of blood transfusion or chronic transfusion therapy is not recommended for prevention or treatment of VOC or recurrent SCD pain.
Red blood cell (RBC) alloimmunization occurs in approximately 30% of transfused sickle cell disease patients compared to 2% to 5% of all transfusion recipients.47
In the severest cases, hyperhemolysis, defined by the destruction of both transfused and autologous RBCs, occurs and may be life-threatening.
Blood transfusion may be indicated where all other measures have failed and for reasons other than pain (eg, stroke, silent stroke, abnormal transcranial Doppler ultrasound, or pregnancy).
Where indicated, blood transfusion aims to increase the oxygen-carrying capacity of blood and to decrease the proportion of sickle hemoglobin (HbS) relative to hemoglobin A (HbA).
In the acute situation, simple transfusion will increase oxygen-carrying capacity but with a risk of hyperviscosity if the Hb is increased to significantly over the patient’s baseline. Therefore, the target Hb should be 10 g/dL in patients with homozygous HbS (HbSS) 54. Exchange transfusion has the advantage of both increasing oxygen-carrying capacity and reducing HbS percentage.
Other Preventive Measures
• prophylactic administration of penicillin in childhood (vaccine)
• avoiding temperature extremes
• good hydration
• antimalaria prophylaxis
RECOMMENDATIONS FOR MANAGING A SICKLE CELL CRISIS REMOTELY, AT THE PATIENT’S HOME
Management of sickle cell crisis requires immediate termination; the longer the crisis goes on, the greater the ischemia, inflammatory storm, and multi-organ damage. Prolonged severe pain crises perpetuate a vicious cycle with chest-splinting from pain, increased hypoxia leading to increased vaso-occlusion, increased inflammation, and increased pain.
Once a patient enters a full-blown crisis, administration of oxygen alone will not abort it, and if the pain crisis is not controlled within the first 30 minutes, it will progress and the patient will require hospitalization for days to weeks to control the crisis.
.
Actively Treating a Sickle Cell Pain Crisis
When a sickle cell crisis presents, time is of the essence to interrupt the pain, terminate the neuro-inflammatory cascade, and prevent organ damage. Pain is more than just a symptom but is indicative of vaso-occlusion, ischemia, and multi-organ damage.
The longer the interval to treatment, the greater the ischemic organ damage, the greater the amplification of the inflammatory cascade, and the more protracted the crisis becomes. The 2020 guidelines for the management of acute and chronic pain by the American Society of Hematologists recommend assessment and administration of analgesia within 1 hour of arrival to the ER.37 Taking into consideration the time for transportation to the hospital, this is too long. It is important to realize that a sickle cell pain crisis represents much more than severe excruciating pain. It represents an inflammatory storm, ongoing ischemia, and multi-organ damage.
The following sections address treatment recommendations whether the patient is at home with remote monitoring by a clinician or clinical team, or in the hospital
Sickle cell anemia patients should be trained along with their relatives or caregivers. They should have available at home the following equipment set up to enable safe and timely administration of the opioid and anti-inflammatory medications.
- Oxygen cylinder or oxygen concentrator
- Equipment for on site or remote vital sign monitoring including oxygen saturation, temperature, respiratory rate, blood pressure
- Intranasal naloxone
- Remote video for monitoring by the physician or monitoring station
- Unit dose of a parenteral opioid eg Dilaudid that the patient has tolerated before
- Unit dose of a parenteral NSAID eg Ketorolac that the patient has tolerated before
RECOMMENDATIONS FOR THE MANAGEMENT OF A SICKLE CELL CRISIS IN THE HOSPITAL OR HEALTHCARE FACILITY
Should the pain crisis fail to resolve or there are additional complications, including fever, chills, shortness of breath, increased redness of the urine signifying hemolysis, chest or abdominal pain, the patient should be advised to proceed to the hospital and seek immediate medical attention.
In the medical clinic or hospital, the patient should be evaluated for causes of pain other than a VOC. If the VOC pain is atypical, the medical team should evaluate for other possible etiologies of pain.
A history and physical examination and diagnostic tests should be performed. These should include a complete blood count (CBC) with differential, reticulocyte count, serial CBC, chest x-ray or CT scan, and in patients with a temperature > 101.3°F (38.5°C), blood culture and urine culture where urinary tract infection is suspected, and blood smears for malarial parasites, where malaria is suspected.68
Pain and inflammation control by the parenteral route in combination with oxygen therapy must be rapidly initiated within the first 30 minutes.
Guidelines for Pain Control as published by the National Heart, Lung and Blood Institute, Expert Panel Report, 2014 Evidence-Based Management of Sickle Cell Disease and incorporating the ASH 2020 Guidelines, include the following:69,70
- Calculate the parenteral (IV or SC) opioid dose based on total daily short-acting opioid dose currently being taken at home to manage the VOC. The ASH guideline panel suggests tailored opioid dosing based on consideration of baseline opioid therapy and prior effective therapy37
- Administer parenteral opioids using the subcutaneous route when intravenous access is difficult.
- Reassess pain and re-administer opioids, if necessary, for continued severe pain every 15 to 30 minutes until pain is under control per patient report.
- Maintain or consider escalation of the dose by 25% until pain is controlled.
- Reassess after each dose for pain relief and side effects.
- Opioid sparing medication such as sub-anesthetic ketamine 5 mg to 7.5 mg SQ/IM/IV (children 0.1 mg/kg) may be utilized to decrease the amount of opioids required as well as decrease the side effects, while providing effective pain control. (Author’s Note:ASH dosing recommendation for ketamine infusion for pain is weight-based at 0.1 mg/kg to 0.3 mg/kg/hour with a max of 1 mg/kg/hour. The author has found the higher limit to be too high and administers ketamine daily for pain procedures to adults using single hourly doses of 5 mg to 7.5 mg (0.1-0.15 mls) with good results.)
- To reduce the risk of acute chest syndrome in adults and children hospitalized for a VOC, encourage use of incentive spirometry every 1 to 2 hours, while awake.
- In euvolemic adults and children with SCD and a VOC who are unable to drink fluids, provide intravenous hydration at no more than maintenance rate to avoid over-hydration. Except where there is clinically significant dehydration, IV fluids should be administered cautiously as there is risk of harm in patients with cardiopulmonary compromise.
- In adults and children with SCD and a VOC being treated with opioids, monitor for excessive sedation by measuring sedation with an objective sedation scale and oxygenation levels.
- Gradually titrate down parenteral opioids as VOC resolves.
- In adults and children with SCD and a VOC, do not administer a blood transfusion unless there are other indications for transfusion, such as: simple transfusion for symptomatic ACS combined with a decreased Hb of 1g/dl below the baseline; exchange transfusion for symptomatic severe ACS (as defined by an oxygen saturation less than 90%); despite supplemental oxygen; simple transfusion for acute splenic sequestration plus severe anemia; and simple or exchange blood transfusion for stroke.
Subsequent treatment in patients with fever should include intravenous antibiotics that provide coverage against community-acquired pneumonia and gram-negative enteric organisms, treatment for malaria as indicated, as well as simple or exchange blood transfusion where indicated. Assess patients whose hemoglobin concentration is 2 g/dL or more below their baseline (or less than 6 g/dL when the baseline is unknown) for acute splenic sequestration, anaplastic episode, a delayed hemolytic transfusion reaction, ACS, and infection.
Clinicians can manage aplastic events with immediate red blood cell transfusion aimed at restoring the hemoglobin to a safe (not necessarily baseline) value. Isolation of hospitalized patients (droplet precautions) is required to prevent the spread of the parvovirus B19 to pregnant women and others with SCD or compromised immunity. In people with hypovolemia due to severe acute splenic sequestration, immediately provide IV fluid resuscitation
References:
- https://www.researchgate.net/publication/351357496_Pain_Crises_in_Sickle_Cell_Disease_A_Clinical_Guide_to_Prevention_and_Treatment_How_to_Manage_an_Acute_Pain_Crisis_in_Sickle_Cell_Disease_Practical_Recommendations
- https://www.researchgate.net/publication/338984448_Sickle_Cell_Pain_Crisis_Clinical_Guidelines_for_the_Use_of_Oxygen
- https://www.practicalpainmanagement.com/pain/other/how-manage-acute-pain-crisis-sickle-cell-disease-practical-recommendations


Responses